
Impairment of PI3K/Akt Signaling Pathway in Brain in Alzheimer’s Disease¶
Foreword¶
For the past few years, I have occasionally studied and participated in research related to functional genetics in Alzheimer’s disease. Functional genetics strives to understand the relationship between genotype and phenotype on a genome-wide scale. Such studies employ a multi-omic approach to answer multitude of questions relevant to disease formation and development on a genetic level. Since most Mendelian (single gene variation derived) diseases are already well known, much of the research in functional genetics is focused on multifactorial (complex) diseases, which develop from multiple different genetic variants and environmental factors. Since genes and their products are almost always cross-interacting, it is important to map the interaction networks to understand what mechanisms underlie within complex diseases. To map these networks, one must first detect the mutations and understand gene product interaction pathways with traditional laboratory research. However, to focus on specific mutations and pathways can lead to missing the forest for the trees. Once enough data is accumulated, it can be interpreted with bioinformatic approaches to discover new contributing factors and larger networks that affect the development of certain complex disease, or even connect certain diseases together in a cause-and-effect fashion, giving another dimension to functional genetics.
I will use an English draft of my bachelor’s thesis as a background for my personal bioinformatic experiments with Alzheimer’s disease related open data, that I will conduct in the future. Since this is just an early draft, it does not fully represent my bachelor’s thesis, nor is it a proper peer reviewed scientific article. However, you may find some interesting and relevant information therein related to molecular mechanisms in Alzheimer’s disease, and understand my rationale for more pro-active approaches to treat complex diseases.
Abstract¶
Changes in PI3K/Akt signaling pathway have been discovered in brains of patients with Alzheimer’s disease (AD). Furthermore, insulin/PI3K/Akt signaling pathway is a major factor in type 2 diabetes (T2D) pathology. AD and T2D occur more and more often in aging populations. T2D is a comorbidity factor and a risk factor to AD, increasing the risk of developing AD from two- to four-fold. However, there is not a complete and concise understanding, how the changes in PI3K/Akt signaling pathway affect central nervous system (CNS). In periphery, insulin can act as growth factor, and alter both glucose, fatty acid, and protein metabolism, where PI3K/Akt signaling pathway is the pivotal mediator of these effects. In CNS, insulin, via PI3K/Akt pathway, acts as important neuroprotective agent. Activation of PI3K/Akt signaling pathway in CNS, by insulin treatment, improved memory in both mouse and human experiments. In this review, we will examine literature dealing with PI3K/Akt signaling pathway’s features, mechanisms, and significance to CNS’s function. Also, we will review intranasal insulin treatment’s influence on PI3K/Akt signaling pathway in brain.
Contents¶
- Abstract
- Introduction
- 1 Composition of PI3K/Akt signaling pathway
- 2 Detrimental factors for PI3K/Akt signaling pathway
- 3 How AD pathogenesis alters PI3K/Akt signaling pathway in brain
- 3.1 Impairment of insulin signaling and PI3K/Akt pathway
- 3.2 Effect of hyperphosphorylated tau on PI3K/Akt pathway
- 3.3 Effect of Akt inactivity on other cellular metabolic pathways
- 4 Effect of intranasal insulin treatment on PI3K/Akt signaling pathway in brain
- 5 Discussion
- 6 Sources
Introduction¶
In this review we shall investigate the role and effects of the impairment of PI3K/Akt signaling pathway in Alzheimer’s disease. Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) is a kinase family involved in several intracellular signaling transduction processes (Fruman, Meyers and Cantley, 1998). PI3K relays phosphoinositide signaling by converting phosphatidylinositol (3,4)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3). These phosphoinositide pathways end with a variety of kinases, such as Akt, that plays a key role in cell growth and metabolism (Lodish et al., 2016). Akt is a serine/threonine-protein kinase that upon activation through phosphorylation by phosphoinositide signaling, phosphorylates and inactivates pro-apoptotic proteins and promotes survival by phosphorylating serine/threonine residues of pro-survival transcription factors, such as Forkhead related ligand (FKHRL) and Forkhead box (FOX) and by preventing release of cytochrome c (Noguchi and Suizu, 2012).
Alzheimer’s disease (AD) is a neurodegenerative disease, characterized by aggregates of amyloid-beta (Aβ) in synaptic clefts, accumulation of hyperphosphorylated tau as neurofibrillary tangles (NFTs) in neuronal axons, changes in insulin signaling in central nervous system (CNS) and neuronal loss. The amyloid cascade hypothesis has been a well-established explanation of the etiology and pathogenesis of AD. It proposes that the deposition of Aβ is the initial pathological event in AD leading to the formation of senile plaques (SPs) and then to NFTs, neuronal cell death, and ultimately dementia. While there is substantial evidence supporting the hypothesis, SPs and NFTs may develop independently, and they may be the products rather than the causes of neurodegeneration in AD (Reitz, 2012).
Altered insulin signaling in CNS especially has major consequences in nerve cell’s intracellular PI3K/Akt signaling pathway, due to activation of PI3K/Akt signaling pathway being reliant on activation and dimerization of insulin receptor (IR). Impairment of the PI3K/Akt signaling pathway leads to diminished inhibition of glycogen synthase kinase 3 beta (GSK3β), which leads to accumulation of hyperphosphorylated tau in neural cell axons, forming NFTs and thus further promoting the development of AD.
This subject was chosen due to relationship between PI3K/Akt signaling pathway and AD not having enough academic analysis in the field of molecular biology and the subject of AD itself is still relevant. The goal of this study is to collect information about the detrimental factors for PI3K/Akt signaling pathway and the effects of impaired PI3K/Akt signaling pathway in AD and form conclusions of collected data.
The concepts of this study will be narrowed down to PI3K/Akt signaling pathway in various cells (such as astrocytes and oligodendrocytes) of CNS in context of Alzheimer’s disease. We will investigate how certain metabolic diseases, such as type 2 diabetes (T2D), mutations in Akt2 and how dysfunctional insulin signaling in CNS will affect PI3K/Akt signaling pathway and how this will further affect AD at cellular and molecular level. Lastly, we will investigate certain therapeutic methods focusing on PI3K/Akt signaling pathway.
1 Composition of PI3K/Akt signaling pathway¶
1.1 PI3K¶
PI3K is a family of protein kinases divided into three classes: Class I, class II and class III (Fruman, Meyers et al. 1998). These three classes of PI3k are divided in to isoforms: class I PI3K is divided into four isoforms, class II PI3K is divided in to three isoforms and class III has only one isoform. PI3K regulates intracellular metabolic signaling pathways, regulates several inflammation responses (Hawkins and Stephens, 2015).
1.1.1 Structure of PI3K¶
Class I PI3Ks consist of a p110 catalytic subunit, of which there exists three variants: p110α, p110β, and p110δ (figure 1.), and of a p85 regulatory subunit of which there exists five variants: p50α, p55α, p55γ, p85α, and p85β (figure 1.). Class I may consist of a p110γ catalytic subunit with a p87 or p101 regulatory subunit, but these lack SH2 domains, have no homology to other proteins and have no identifiable domains. P85 subunits contain Src homology domain 2 and 3 (SH2 and SH3). Class II PI3Ks consist of three isoforms: PI3K-C2α, PI3K-C2β and PI3K-C2γ. Class II PI3Ks lack regulatory subunits but have amino‑ and carboxy‑terminal extensions to the PI3K core structure, which could mediate protein–protein interactions. Class II PI3Ks also have specific lipid binding phox homology (PX) domains. Class III PI3K has one catalytic member, vacuolar protein sorting 34 (Vps34). Vps15 consists of a catalytic domain, HEAT domains, which possibly mediate protein–protein interactions and WD repeats, which are essential for interaction with other proteins. All PI3K catalytic subunits have a PI3K core structure consisting of a C2 domain, a helical domain and a catalytic domain (Vanhaesebroeck et al., 2010).
SH2 domain acts as a guide that leads the PI3K to the plasma membrane by binding to phosphotyrosines on receptor tyrosine kinases (RTKs) cytosolic domain. PI3Ks p110 adds phosphate to the 3’ carbon in the phosphaditylinositol-4,5-biphosphate (PIP2) forming phosphaditylinositol-3,4,5-triphosphate (PIP3) whilst depleting a single ATP into ADP (figure 4.) (Lodish et al., 2016).
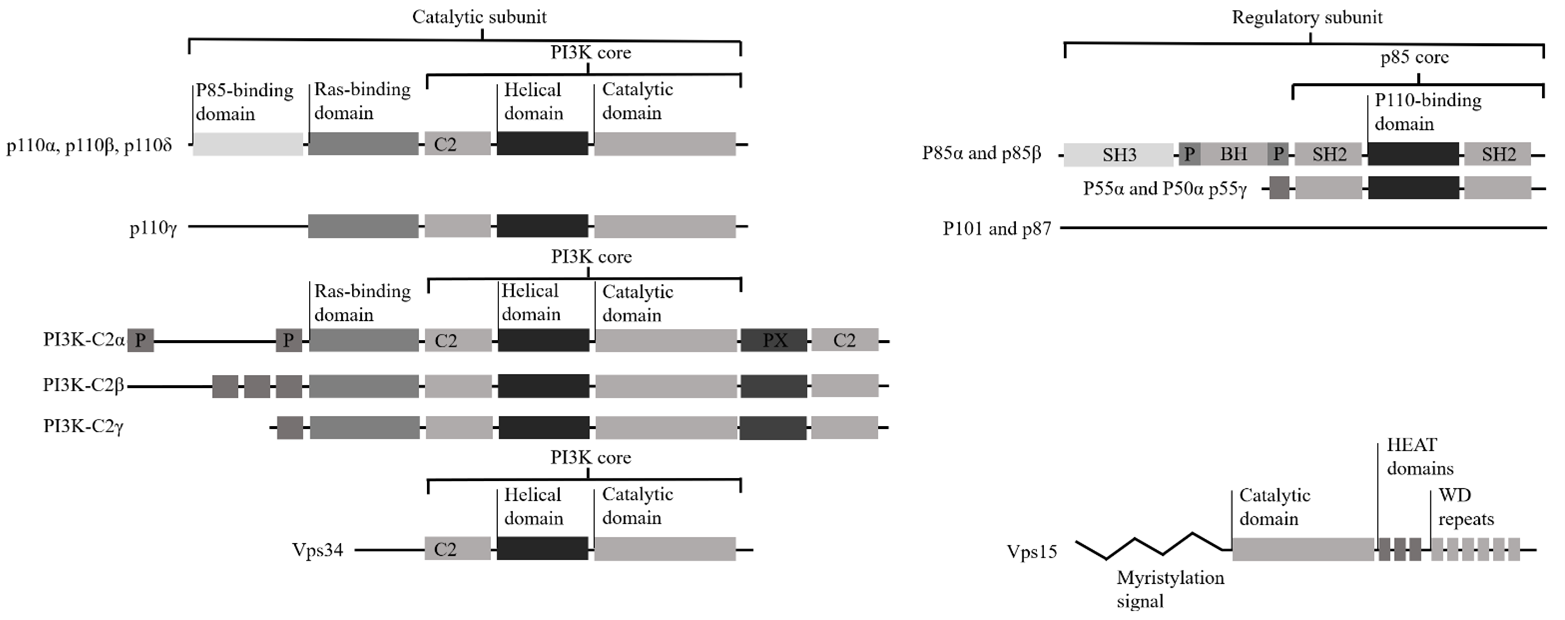
Figure 1. Structure of PI3K. Class I PI3Ks consist of a p110 catalytic subunit, of which there exists three variants: p110α, p110β, and p110δ, and of a p85 regulatory subunit of which there exists five variants: p50α, p55α, p55γ, p85α, and p85β. Class I may consist of a p110γ catalytic subunit with a p87 or p101 regulatory subunit, but these lack SH2 domains, have no homology to other proteins and have no identifiable domains. P85 subunits contain Src homology domain 2 and 3 (SH2 and SH3). Class II PI3Ks consist of three isoforms: PI3K-C2α, PI3K-C2β and PI3K-C2γ. Class II PI3Ks lack regulatory subunits but have amino‑ and carboxy‑terminal extensions to the PI3K core structure, which could mediate protein–protein interactions. Class II PI3Ks also have specific lipid binding phox homology (PX) domains. Class III PI3K has one catalytic member, vacuolar protein sorting 34 (Vps34). Vps15 consists of a catalytic domain, HEAT domains, which possibly mediate protein–protein interactions and WD repeats, which are essential for interaction with other proteins. All PI3K catalytic subunits have a PI3K core structure consisting of a C2 domain, a helical domain and a catalytic domain (Edited from Vanhaesebroeck et al., 2010).
1.1.2 Activators of PI3K¶
Activated RTKs, such as insulin receptors (IR), and cytokine receptors initiate another phosphoinositide pathway by recruiting the PI3K to the membrane. PI3K is recruited to the plasma membrane by binding of its SH2 domain to phosphotyrosines on the cytosolic domain of many activated RTKs and cytokine receptors (Lodish et al., 2016). The IR and IGF-1R, major mediators of PI3K activity, have a heterotetrameric structure with extracellular localized α-subunits and membrane bound β-subunits. These β-subunits contain ATP-binding motifs, autophosphorylation sites, and tyrosine protein kinase activity activated after binding of insulin or IGF-1 to the receptor (Moll and Schubert, 2012; Arnold et al., 2018).
Binding of the ligand (such as insulin, or IGF) leads to a conformational change of the IR, activating its tyrosine kinase activity. This activity leads to recruitment and phosphorylation of downstream signaling proteins such as insulin receptor substrate (IRS) family of proteins. The IRS protein family consists of four members, IRS1, IRS2, IRS3 and IRS4, (Moll and Schubert, 2012). IRS1 and IRS2 are the best characterized, most widely distributed and most relevant to the classic metabolic actions of insulin. Although IRS1 and IRS2 have overlapping signal transduction activity, IRS1 is especially important in skeletal muscle, adipose tissue and the cerebral cortex whereas IRS2 is important in the liver and hypothalamus (Talbot et al., 2012).
These IRS proteins consist of a pleckstrin homology (PH) domain located at the N-terminus, a phosphotyrosine binding (PTB) domain and a C-terminus with multiple tyrosine phosphorylation sites. These phosphotyrosine motifs of the IRS proteins are binding sites for Src homology (SH)2 domain-containing proteins (Moll and Schubert, 2012). IRS1 and IRS2 activity is regulated by feedback of downstream kinases Akt, GSK3β, mTOR and MAPK, through site-specific serine phosphorylation (Arnold et al., 2018).
1.1.3 Substrates and products of PI3K¶
Phosphorylation on the 3rd position of inositol in PIP2 yields the second messenger PIP3. Target for this second messenger is a protein-serine/threonine kinase Akt which has PIP3 binding PH domain. As the PIP3 binds to the Akt it is recruited to the inner face of the plasma membrane where it is phosphorylated and activated by PDK1, which contains the PH domain as well. Akt requires phosphorylation at a second site by a protein kinase mTORC2 to be activated (Cooper and Hausman., 2009).
1.2 Akt¶
Phosphoinositide pathways end with a variety of kinases, including serine/threonine kinase Akt. Akt consists of three isoforms: Akt1, Akt2 and Akt3. This family of protein kinases, once activated, regulates cell survival, proliferation, cell cycle, cytoskeletal organization, cell metabolism, angiogenesis, vesicle trafficking, glucose transport, cell growth and cell metabolism (Noguchi and Suizu, 2012; Cohen, 2013). Also, Akt signaling is important for mediating oligodendrocyte proliferation, survival, differentiation and myelination (Ye, P. et al., 2002).
Akt1 and Akt3 are distributed throughout somatic layers of the hippocampus, with varying local expression. Akt1 expresses more intense immunoreactivity in the cell body layer area, stratum pyramidale of CA1, whereas Akt3 expresses more intense immunoreactivity in cell bodies within area of CA3 and the hilus of the dentate gyrus but has reduced expression in the CA2/CA1 region. Akt2 is mostly expressed in cells of the molecular layer of the dentate gyrus and stratum radiatum of CA1 and may solely be expressed in astrocytes and not in neurons of the hippocampus (Levenga et al., 2017).
Akt is involved in insulin mediated regulation of glucose homeostasis and is closely linked to T2D (Cohen, 2013). Akt also is a key mediator of GSK3β (Leibrock et al., 2013). PIP3 recruits PDK1 to the plasma membrane, which in turn partially activates Akt by phosphorylating its T308 residue. Full activation of Akt requires phosphorylation its S473 residue by mammalian target of rapamycin complex 2 (mTORC2) (figure 4.) (Cooper and Hausman 2009).
1.2.1 Structure of Akt¶
Akt activation is regulated by phosphorylation at two amino acid residues: threonine 308/309/305 and serine 473/474/472 (Akt1/2/3 respectively) (Noguchi and Suizu, 2012; Cohen, 2013). All three Akt isoforms bear a highly conserved pleckstrin homology (PH) domain which binds to PIP3. These isoforms also contain a conserved PKC-like kinase domain at the centre of the molecules and regulatory domain present at the C-terminal end. Activation of Akt is triggered by interacting with PIP3 with its Variable Loop 1 (VL1) of the PH-domain, which induces conformational changes, permitting phosphoinositide-dependent protein kinase 1 (PDK 1) to access to the threonine at 308/309/305 of Akt at the plasma membrane (figure 2.) (Noguchi and Suizu, 2012).
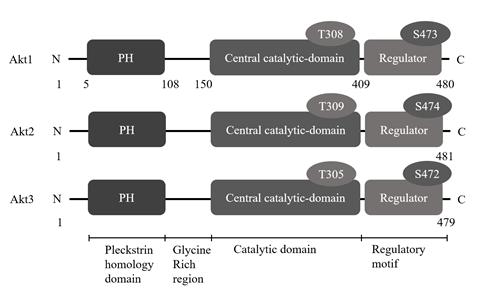
Figure 2. Structures of Akt isoforms. All three Akt isoforms bear a highly conserved PH domain which binds to PIP3. These isoforms also contain a conserved PKC-like kinase domain at the centre of the molecules and regulatory domain present at the C-terminal end. Activation of Akt is triggered by interacting with PIP3 with its VL1 of the PH-domain, which induces conformational changes, permitting PDK1 to access to the threonine at 308/309/305 of Akt at the plasma membrane (edited from Cohen, 2013).
1.2.2 Modulators of Akt¶
PIP3 has a high affinity for the PH domain of Akt. Binding of PIP3 and PH domain alters the conformation of the PH domain and allows phosphorylation of the T308/309/305 residue of Akt. The binding also leads to recruitment of Akt to the plasma membrane (Noguchi and Suizu, 2012).
Phosphorylation two residues: T308/309/305 and S473/474/472 of Akt (Akt1/2/3 respectively) are identified as governing modulators of Akt activation. T308 is activated by PDK1 at the plasma membrane. Another phosphatase that modulates Akt activity is PTEN, which is a lipid phosphatase that counter-regulates PI3K and its shown to be inversely correlated with Akt2 phosphorylation on S474. Phosphatases that phosphorylate S473/474/472 are referred as PDK2. It has been identified that mTORC2 is the mainstream kinase that specifically phosphorylates S473/474/472 (Noguchi and Suizu, 2012).
1.2.3 Substrates and products of Akt¶
Akt is a cell survival factor exerting anti-apoptotic activity by preventing release of cytochrome c from mitochondria and inactivating forkhead-related ligand (FKHRL) and Forkhead box protein O (FOXO) (Noguchi and Suizu, 2012). In many cells, activated Akt directly phosphorylates and inactivates pro-apoptotic proteins such as Bad, a short-term effect that prevents activation of an apoptotic pathway leading to cell death. Activated Akt also promotes survival of many cultured cells by phosphorylating the Forkhead transcription factor FOX03a on multiple serine/threonine residues, thereby reducing its ability to induce expression of several pro-apoptotic genes (Cooper and Hausman 2009). Akt also modulates a certain protein kinase of special interest for our topic: GSK3β (Thotala and Yazlovitskaya, 2011).
1.3 GSK3β¶
GSK3β is a serine-threonine kinase belonging to the glycogen synthase kinase subfamily and acts as a negative regulator of glucose homeostasis by its ability to phosphorylate and inactivate glycogen synthase. GSK3β is involved in energy metabolism, inflammation, ER-stress, mitochondrial dysfunction and apoptotic pathways. GSK3β is expressed in almost all the tissues. However, abundant expression is detected in brain tissue, especially in the neurons when compared to the astrocytes. The high level of expression in the brain is due to its vital role in the neuronal signaling (Thotala and Yazlovitskaya, 2011).
GSK3β is a multifunctional protein kinase and is regulated by inhibitory phosphorylation of its S9 residue (figure 3.) and activating phosphorylation of its Y216 residue (figure 3.) (Thotala and Yazlovitskaya, 2011). GSK3β has important effect on Wnt signaling pathway. GSK3β can phosphorylate β-catenin to induce its degradation by the proteasome. β-catenin plays an important role in neurogenesis and neural differentiation, through Wnt/β-catenin pathway (Zheng et al., 2017). GSK3β is reported to regulate by phosphorylation over 40 different proteins, such as glycogen synthase, Acetyl CoA carboxylase, Cyclin D1, p53, microtubule associated proteins (MAPs), which includes tau (Thotala and Yazlovitskaya, 2011; Arnold et al., 2018). Through these diverse proteins, insulin and GSK3β signaling play important parts in the regulation of cellular proliferation, migration, glucose regulation, apoptosis and neuroplasticity (Arnold et al., 2018).
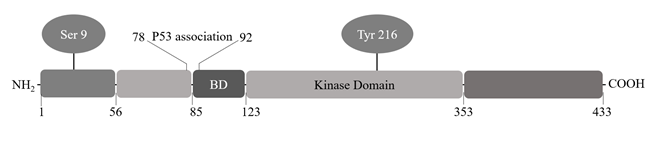
Figure 3. Structure of GSK3β. GSK3β is a 46-47 kDa protein consisting of 433 amino acids. The protein contains an N-terminal domain, a kinase domain and a C-terminal domain. Phosphorylation of Tyrosine 216 residue located in the T-loop facilitates substrate phosphorylation by GSK3β but is not strictly required for its kinase activity. Phosphorylation of GSK3β at serine 9 residue in N-terminal region leads to inhibition of its kinase activity. Binding domain (BD) includes GSK3β specific binding sites for substrates and protein complexes (edited from Thotala and Yazlovitskaya, 2011).
1.3.1 hyperphosphorylated tau and neurofibrillary tangles¶
Tau is a MAP, encoded by a single gene, MAPT, which lies on human chromosome 17 (Iqbal, Liu and Gong, 2016). Tau constitutes a family of six isoforms with a range of 352-441 amino acids. Tau acts as a MT stabilizer, giving length and rigidity to axons. Unregulated GSK3β phosphorylates tau, which forms hyperphosphorylated form of tau. Hyperphosphorylated tau occurs at early stage of AD (Kang et al., 2017) and contributes to formation of NFTs and MT destabilization, though it may have cognition impairing effects preceding histologically identifiable NFTs (Kang et al., 2017).
Collapsin response protein-2 (CRMP2) is a member of a separate class of MAPs that is distinct of tau and is indirectly related to MT stabilization. The CRMP2 is target of phosphorylation by GSK3β as well, which in its hyperphosphorylated form acts as a co-aggregate with hyperphosphorylated tau in formation of NFTs. These destabilized MTs in neural axons contribute in formation of dystrophic neurites (Hensley and Kursula, 2016).
NFTs, primarily made up of paired helical filaments (PHFs), are intraneuronal aggregates of hyperphosphorylated and misfolded tau that become extraneuronal when NFT-bearing neurons perish. NFTs have a generic spatiotemporal progression that correlates with the severity of the cognitive decline (Serrano-Pozo et al., 2011). NFT development occurs in three stages divided into preneuro fibrillary tangle, intraneuronal fibrillary tangle and extraneuronal neurofibrillary tangle. Hyperphosphorylation of tau occurs at S396, S404 and T231 in the intraneuronal neurofibrillary tangle stage and at S199, r202 and S409 in the preneuro fibrillary tangle stage. Especially S404, emerging very early in the disease process, is a significant site in a PHF, which is the main fibrous component of NFTs and contains predominantly hyperphosphorylated tau (Kang, Xiang et al. 2017).
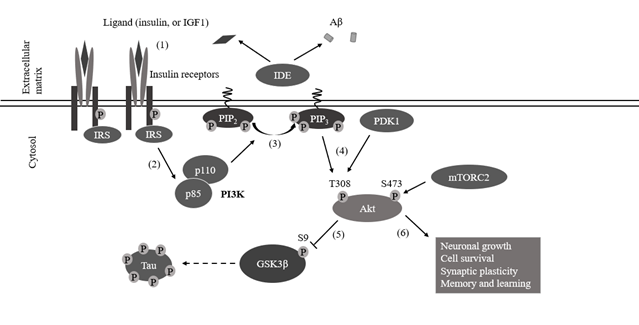
Figure 4. PI3K/Akt signaling pathway. (1) Once binding of the ligand, autophosphorylated β-subunits of insulin receptors recruit insulin receptor substrates (IRSs). The tyrosine kinase activity of insulin receptors phosphorylates tyrosine residues on IRS1 or IRS2, which activates these keystones of insulin action and stimulates signaling via the AKT pathway. (2) IRS recruits PI3Ks p85 subunit by binding to its SH2 domain. (3) PI3Ks p110 domain adds phosphate to the 3’ carbon in the PIP2 producing PIP3. (4) Akt is recruited to the plasma membrane by PIP3 and phosphorylation of its T308 and S473 residues by PDK1 and mTORC2. (5) Akt inhibits GSK3β activity by phosphorylating GSK3β’s S9 residue. (6) Signal pathway activation leads to downstream responses, which induce neuronal growth, cell survival, synaptic plasticity, memory and learning (edited from Yu JS, Cui W., 2016).
2 Detrimental factors for PI3K/Akt signaling pathway¶
2.1 Aβ¶
Amyloid plaques result from the accumulation of the Aβ, which is believed to occur early in the disease progress and initiate downstream events such as tau phosphorylation, inflammation and oxidative stress (Verdile et al., 2015). Studies in human biopsies and experimental animal models have shown that Aβ-induced oxidative stress plays a critical role in hyperphosphorylation of tau, which together drive neurodegeneration and pathogenesis of AD (Verdile et al., 2015; Singh et al., 2017).
APP containing 751 or 770 amino acids (APP751 and APP770) is expressed in non-neuronal tissue, while APP695 is predominantly found in neurons (Kang and Muller-Hill, 1990). APP protein has a large N-terminal extracellular domain, constituting the soluble APPα/β (sAPPα/β) domain, followed by a transmembrane region that contains the Aβ peptide sequence, and a short C-terminal cytoplasmic tail domain, called the APP intracellular domain (AICD) (Reinhard, Hébert and De Strooper, 2005).
APP processing is divided into two pathways: amyloidogenic and non-amyloidogenic. Amyloidogenic APP processing pathway proceeds as proteolytic cleavage of APP is promoted by the β-secretase BACE-1 (β-site APP cleaving enzyme 1). APP is cleaved at the N-terminus via BACE-1 generating soluble APPβ and the C-terminal fragment C99. Cleavage of C99 by γ-secretase produces Aβ and the AICD. Released Aβ-peptides are mainly found in two variants: Aβ40 and Aβ42, of which the Aβ42 is susceptible to aggregate, forming neurotoxic oligomers (Vassar, 2004). In non-amyloidogenic pathway APP is cleaved by α-secretase in the Aβ domain. This produces two fragments, the neuroprotective soluble APPα fragment, and a small carboxy-terminal fragment (C83). C83 is further cleaved by the γ-secretase to generate P3 and AICD (Pearson and Peers, 2006).
The soluble Aβ oligomers upon binding to ephrin type-A receptor 4 RTK trigger abnormal hyperactivity in ionotropic glutamate receptors consequently hampering excitatory synaptic transmission. Thus Aβ-mediated altered activation of these receptors induces Ca2+ influx which leads to long term potentiation, oxidative stress, and eventual apoptotic cell death (Singh et al., 2017). The accumulation of Aβ oligomers can inhibit the autophosphorylation of IRs and reduces their levels and activity within dendrites of hippocampal neurons (Verdile et al., 2015).
The soluble Aβoligomers can inhibit IRs by increasing cytosolic Ca2+, through N-methyl-D-aspartate (NMDA) receptor activation, increasing Akt1 phosphorylation at its S473 residue, which is known to inhibit insulin-induced hippocampal IR pY (Talbot, et al., 2012). Elevated phosphorylation of IRS1 results in inability to transmit signals to PI3K, resulting in failure of PI3K/Akt signaling pathway to mediate downstream pathways such as Wnt/β-catenin pathway, mTOR signaling and GSK3β pathway (Verdile et al., 2015).
The p-PI3K level significantly decreased in the Aβ1-42-treated mice, as well with reduced phosphorylation of Akt at S473 residue and reduced phosphorylation of GSK3β at S9 residue (Ali and Kim, 2015). Aβ1-42 treatment to hippocampal neurons up-regulated expression of p-mTOR and p-PI3K that are known to play important roles during the stressed condition to support survival and growth of neurons. At the same time, Aβ1-42 down-regulated the significant expression and phosphorylation of Akt1 and neurogenesis transcription factor CREB that impairs the proliferation and differentiation of hippocampal neurons and promote neurodegeneration (Singh et al., 2017).
2.2 T2D¶
Insulin resistance, which refers to impaired or failed cell response to insulin receptor activated signaling which results in reduced glucose uptake by the cell, induced by T2D, may be contributing factor to the progression of AD (Li and Hölscher, 2007; Arnold et al., 2018). Insulin regulates blood sugar levels but also acts as a growth factor on all cells including neurons in the CNS. Thus, impairment of insulin signaling affects blood glucose levels but also causes numerous neurodegenerative processes (Li and Hölscher, 2007).
Insulin and its receptors are abundant in hippocampus where insulin signaling is recognised to have function in synaptic plasticity, modulation of learning and memory and has neuroprotective properties. Insulin can promote glucose uptake through regulation of glucose transporter 4 (GLUT4), though it is only expressed in certain neurons, instead the primary glucose transporters in brain are GLUT1 expressed in astrocytes and GLUT3 expressed in neurons (Verdile et al., 2015). GLUT4 is co-expressed with GLUT3 in rodent brain regions related to cognitive behaviours, which include the basal forebrain, hippocampus, amygdala, and, in lesser degrees cerebellum and the cerebral cortex (Apelt, J., Mehlhorn, G. and Schliebs, R. 1999).
Link between T2D and AD can be identified by the metabolic changes in the CNS by chronic inflammation, impaired PI3K/Akt signaling. Furthermore, self-enforcing interplay between AD and T2D affects more general processes such as angiopathic and cytotoxic developments, the induction of apoptosis, or of non-apoptotic cell death via production of free radicals. Insulin resistance refers to impaired or failed cell response to insulin receptor activated signaling which results in reduced glucose uptake by the cell (Li and Hölscher, 2007; Henriksen et al., 2011).
In brain, insulin is known to promote neurite outgrowth, regulate of trafficking of ligand-gated channels, modulate activity dependent synaptic plasticity via PI3K/Akt signaling pathway (Kothari et al., 2017). Insulin and its receptor are abundant in hippocampus and disruption of insulin signaling can have contributing effect to the progression of AD. Elevated phosphorylation of IR serine residues results in inability to transmit signals to PI3K, which has downstream effect of reduced phosphorylation GSK3β S9 residue, leading to loss of regulation of GSK3β and overproduction of hyperphosphorylated tau (figure 5.) (Verdile et al., 2015; Laws et al., 2017). Brain insulin resistance can cause functional detriments, such as impaired ability to regulate brain or periphery metabolism, as well impaired cognition and mood (Arnold et al., 2018).
In a study published by Kothari et al. (2017), they conducted tests on mice by administering the mice diets rich in saturated fat and carbohydrates such as fructose. The results revealed that high fat Western diet + sugar (HFS) on mice resulted in down regulation of Insulin degrading enzyme (IDE), which is responsible for degradation of Aβ. The Aβ aggregates can inhibit the autophosphorylation of IR and even reduce IR levels within dendrites of hippocampal neurons (Henriksen et al., 2011) and thus form a self-promoting neurodegenerative effect. Kothari et al. (2017) also observed increased expression of the active form of GSK3β and hyperactivation of Akt and AMPK S485 in HFS mice compared to control group. Activation of Akt lead to increased synthesis and translocation of glucose transporter from the cytosol to the cell membrane resulting in glucose uptake in brain.
Overall, results by Kothari et al. (2017) demonstrated that HFS decreased tyrosine phosphorylation of IRS1 and increased serine phosphorylation of IRS1, resulting in neuronal insulin resistance. Because insulin is one of the substrates for IDE, decreased levels of insulin are associated with lower IDE expression in brain, which leads to less degradation of Aβ. The HFS diet was found to increase expression of BACE, which can increase APP processing and accumulation of Aβ. Oxidative stress and neuro-inflammation are attributed to central insulin resistance affecting GSK3β levels, which can increase tau hyperphosphorylation. Insulin resistance status is associated with AMPK S485 levels, which can prevent tau dephosphorylation. Increased Aβ toxicity and tau hyperphosphorylation leads to neurodegeneration and decreased synaptic plasticity, a hallmark of AD (Kothari et al., 2017).
Insulin and IGF-1 resistance is most likely a compensatory mechanism to amyloid pathology to reduce Aβ toxicity and promote survival. But decreased IR/IGF-1 signaling causes tau hyperphosphorylation which is neurotoxic at least to certain extend. Experiments in C. elegans suggested that FoxO transcription factors might be involved in mediating the beneficial effects of reduced IR/IGF-1R signaling (Moll and Schubert, 2012).
Alzheimer’s disease Neuroimaging Initiative (ADNI) study showed no association between T2D and accumulation of neocortical Aβ load, nor increases in cerebrospinal fluid (CSF) Aβ42 was associated with lower cortical thickness an increase in CSF total tau and phosphorylated tau. This suggests that T2D may not be associated with cerebral accumulation of Aβ but with other hallmarks of the disease (Laws et al., 2017).
2.3 Hyperphosphorylated tau¶
Talbot et al. (2012) found insulin resistance in the cerebellar cortex, where tau pathology was virtually absent in AD, while Aβ was still present. They also found brain insulin resistance in APP/PS1 mice, which develop major Aβ, but not tau pathology. Conversely, Yarchoan et al. (2014) observed increased levels of hyperphosphorylated tau in high-fat diet (HFD) mice, a model of insulin resistance, providing confirmation that abnormal phosphorylation of IRS1 is a pathological feature of AD and other tauopathies, and provide support for an association between insulin resistance and abnormal tau as well as Aβ (Yarchoan et al., 2014). Serrano-Pozo et al. (2011) found a highly significant, positive correlation between both astrocytosis and microgliosis and NFT burden but not between both reactive glial cell types and amyloid burden, suggesting that glial responses are also related to neurofibrillary degeneration.
2.4 TREM2¶
TREM2 is an immunoreceptor expressed on myeloid cells, including immature dendritic cells, osteoclasts, macrophages, and microglia, and upon activation transmits intracellular signals through its transmembrane binding partner DNAX-activating protein 12 (DAP12) via Src family kinases. DAP12 phosphorylation by TREM2 initiates downstream signaling cascades, which include PI3K, PKC and ERK. TREM2 is specifically expressed in microglia in a healthy brain, whereas TREM2 deficiency has been shown to reduce microgliosis in 5xFADmice with amyloid plaques not fully enclosed by microglia (Zheng et al., 2017).
TREM2 promotes microglial survival by activating the Wnt/β-catenin signaling pathway through posttranslational regulation of β-catenin, which plays essential role in many biological processes during embryonic development and disease pathogenesis, including cell proliferation, survival, migration, and polarity, fate decisions, and self-renewal. Under normal conditions, TREM2 promotes microglial proliferation and survival after injury through the activation of Akt/β-catenin pathways. Knockdown of Trem2 in primary microglia suppresses microglia proliferation. The amount of c-Casp3 was increased, whereas the amounts of pro-Casp3 and the Bcl-2 were decreased in Trem2-/- microglia compared with wild type (WT) microglia suggesting that Trem2 depletion both suppresses microglial proliferation and accelerates microglial apoptosis (Zheng et al., 2017).
PI3K/Akt signaling pathway has been shown to be downstream of TREM2-mediated immune modulation and to regulate β-catenin. TREM2 stabilizes β-catenin by suppressing the proteasomal degradation of β-catenin via Akt/GSK3β signaling pathway. In a study published by H. Zheng et al. (2017) they examined the phosphorylation status of Akt and GSK3β in WT and Trem2-/- microglia, given that phosphorylation of GSK3β at the S9 residue leads to GSK3β inactivation. Akt S473 and GSK3β S9 was decreased significantly in Trem2 knock-down microglia and in Trem2-/- microglia.
2.5 DAM¶
Microglia represent innate immune cells of the CNS that play key roles in neurodegenerative diseases including AD (Rangaraju et al., 2018). Disease associated microglia (DAM) are subset of CNS resident macrophages present at sites of neurodegeneration and is a common signature of microglial response to CNS pathology. DAM show a unique transcriptional and proteomic signature and are characterized molecularly as immune cells expressing typical microglial markers Iba1, Cst3 and Hexb, coincident with downregulation of ‘‘homeostatic’’ microglial genes, including P2ry12, P2ry13, Cx3cr1, CD33, and Tmem119 (Asai et al., 2015). DAM localized with Aβ plaques in the cortex and were absent where Aβ was not present. DAM differentiation is a two-step process where in stage 1 DAM transition is required for further activation of the stage 2 DAM. DAM 1 differentiation occurs Triggering Receptor Expressed on Myeloid cells 2 (TREM2) independently but stage 2 requires TREM2 signal to activate (Deczkowska et al., 2018). TREM2 has been shown to have pro-proliferative and pro-survival function through PI3K, β-catenin and mTOR pathways (Deczkowska et al., 2018).
It is implied that Aβ induces protective DAM early on during disease progression, whereas at late stages, when the accumulation of plaques activates the inflammasome in microglia, DAM become dysregulated and accelerate the disease. This hypothesis is supported by a study published Jay et al., (2017), which showed disease stage-dependent effect on TREM2 deletion on Aβ deposition and inflammatory condition in the brain. In vitro studies of human microglial cultures have showed that microglia express insulin receptors and IRS1 and that insulin modulates microglial inflammatory responses in a complex manner. Depending on insulin concentration in culture, the secretion of certain inflammatory cytokines can be enhanced, and the production of others can be inhibited (Spielman, L. et al., 2015). Importantly AD mouse model experiments showed that DAM have a tendency to locate in CNS regions affected by AD, but not in other regions. DAM co-localized with Aβ plaques in the cortex and were absent from the cerebellum, where Aβ was absent (Deczkowska et al., 2018).
Neuroinflammation refers to the presence of activated microglia and astrocytes, which cause injury through expression and release of pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6 and interferon-gamma (IFNγ). Also, macrophage migration inhibitory factor near Aβ42 plaques supports the concept that neuro-inflammation is an important mediator or propagator of AD neurodegeneration (Mehlhorn G, Hollborn M, Schliebs R., 2000). Neuroinflammation exacerbates insulin resistance, neurotoxic and oxidation-mediated cell death, gliosis, Aβ42 toxicity, and PHF pathology means that there is high association between insulin resistance and neuro-inflammation, though on the other hand these two factors may co-conspire in positive feedback loop to mediate neurodegeneration (de la Monte 2017).
2.6 Akt2 variant P50T¶
P50T is a mutation in Akt2-gene where in the gene product of Akt2, 50th amino acid, proline has switched into threonine, which results in partial loss of Akt2 (Latva-Rasku et al., 2018). In a research article published by Latva-Rasku et al. (2018), a study was conducted where two groups of men, one group of twenty P50T/Akt2 carriers and another group of twenty-five noncarriers were compared in fasting insulin. Participants of the study were matched in age, BMI and fasting glucose. In results of the study the fasting insulin of the P50T/Akt2 carriers was higher compared to the noncarriers. The glucose uptake in the brain was overall higher for the P50T/Akt2 carriers compared to noncarriers. Fasting free fatty acid levels did not differ between sample groups.
3 How AD pathogenesis alters PI3K/Akt signaling pathway in brain¶
3.1 Impairment of insulin signaling and PI3K/Akt pathway¶
AD brain is insulin and IGF1 resistant, though brain IGF-1 resistance in AD may be adaptive, due to deficient IGF-1 signaling delays Aβ accumulation and toxicity in animal models of AD. In AD case studies conducted by Talbot et al. (2012), the cerebellar cortex and, to a greater degree, the hippocampal formation (CA1-CA3, dentate gyrus and the subiculum) showed reduced responses to 1 nM insulin at all levels of the signaling pathway investigated. The reduction in the kinase domain implies downregulation of all insulin signaling pathways, and not just in the IR/PI3K/Akt-pathway (Talbot et al. 2012).
Talbot et al. (2012) concluded that their findings are inconsistent with GSK hypothesis of AD, which asserts that GSK3 is overactive and is accountable for AD pathology, including tau hyperphosphorylation and resulting NFT formation. Their results showed no increase in total GSK3. The hippocampal formation (HF) in AD displayed normal basal levels of GSK3β pS9, but the net effect may be the same as in insulin-resistant skeletal muscle, given the reduced GSK3β pY216 levels observed. CA1 in the HF of AD cases did show elevated basal levels of GSK3α/β pS21/9 despite lower total GSK3β, but that again suggests reduced basal activation of GSK3. While 1 nM insulin stimulation in AD evoked lower levels of GSK-3 β pS9, as in insulin-resistant skeletal muscle, it also evoked lower levels of GSK3β pY216. The net effect of insulin on GSK3β activity in AD is thus unclear. However, contrary to the GSK hypothesis of AD, levels of suppressed GSK3 in CA1 were positively, not negatively, correlated with the density of NFTs detected with an antibody to hyperphosphorylated tau. This may indicate that brain insulin resistance doesn’t significantly affect GSK3-mediated Tau phosphorylation and NFT formation.
3.2 Effect of hyperphosphorylated tau on PI3K/Akt pathway¶
GSK3β hyperactivity, result from impaired PI3K/Akt signaling, as stated earlier is a major factor in production of hyperphosphorylated tau. Hyperphosphorylated tau plays a central role in neurofibrillary degeneration, that is a leading cause of neuronal death in AD. Hyperphosphorylated tau not only loses its biological activity and disassociates from MTs, but it promotes MT polymerization as well. The soluble hyperphosphorylated tau are neurotoxic possibly due to defence mechanisms of the neuron. Hyperphosphorylated tau polymerizes into highly aggregated PHFs (Gong and Iqbal, 2008), which manifest as NFTs in the neuronal soma, as neuropil threads in neurites, and as dystrophic neurites surrounding an amyloid-β (Aβ) core in neuritic plaques. NFTs are one of the two main histopathological hallmarks of AD along with Aβ plaques, and their density directly correlates with disease severity and dementia.
3.3 Effect of Akt inactivity on other cellular metabolic pathways¶
It has been implicated that each Akt isoform is involved in distinct neurological disorders, of which Akt1 mutations have been associated with schizophrenia, Akt2 with gliomas and Akt3 with brain growth (Levenga et al., 2017).
Animal studies on Akt knock out scenarios have revealed, that Akt may play a vital role in cognition and metabolic disorders in brain. In a study published by Leibrock et al. (2013) series of behavioural tests were conducted to a cohort of mice of which consisted of Akt2 knock out and WT mice. Akt2 knock out mice fared worse in every conducted behaviour test, such as Forced Swimming tests, open-field tests and Light Dark Box tests. Akt2 knock out mice showed more anxious and depressive behaviour and were cognitively impaired compared to WT mice.
Isoform-specific roles for Akt have been associated with different disease pathologies. The Akt2 isoform, for example, has been implicated in diabetes. Akt2-deficient mice have resistance to insulin and a subset develop diabetes-mellitus-like syndrome. Akt3 KO mice showed reduced hippocampal levels of GSK3β and Akt3 KO mice had significantly smaller brains. On the other hand, the Akt1 KO mice had normal GSK3β levels (Levenga et al., 2017).
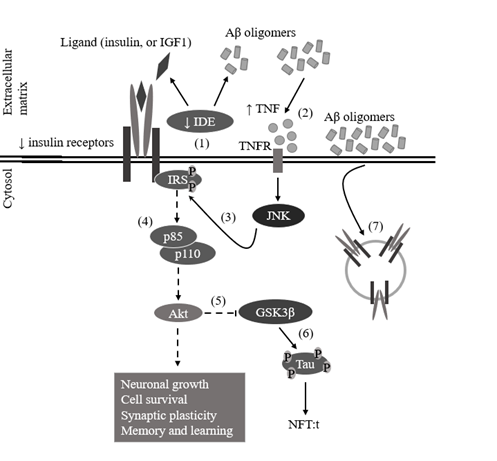
Figure 5. Impairment of PI3K/Akt signaling pathway due to impairment of insulin signaling. (1) Decreased expression of insulin degrading enzyme (IDE) decreases the clearance of Aβ oligomers. (2) Accumulation of Aβ oligomers leads to increased tumor necrosis factor (TNF)-levels, which through TNF receptor (TNFR) activates stress kinases, such as c-Jun N-terminal kinase (JNK). (3) Activated JNK inhibits insulin receptor substrate (IRS) function by phosphorylating their serine residues. (4) Therefore, Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) cannot be recuited and its p110 subunit will not phosphorylate phosphatidylinositol (3,4)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3), leaving Akt inactive. (5) Because Akt is inactive, glycogen synthase kinase 3 beta’s (GSK3β’s) serine 9 (S9) residue will not be phosphorylated, decreasing inhibition of GSK3β activity. (6) Uninhibited GSK3β phosphorylates tau protein, producing hyperphosphorylated tau, which begins to form paired helical filaments (PHFs), which will form neurofibrillary tangles (NFTs). (7) Accumulation of Aβ oligomers around cell membrane’s surface, leads to intake of insulin receptors (IRs) through casein kinase 2 (CK2) and Ca2+/calmodulin dependent kinase II (CaMKII) (edited from Bedse et al., 2015).
4 Effect of intranasal insulin treatment on PI3K/Akt signaling pathway in brain¶
It is hypothesized that brain insulin signaling relates to pathogenesis of AD and that brain insulin resistance and AD share several common traits such as energy metabolic dysfunction, insulin resistance, inflammation and mitochondrial dysfunction. Several studies have established that intranasal insulin treatment have shown positive outcomes in AD cases, both in animal and human experiments.
Increase in PI3K activity by insulin treatment was measured by phosphorylation of its downstream target Akt on S473 using western blot analysis, which was markedly increased. From this, it was concluded that insulin is linked with PI3K activation. Additionally, by treating hippocampal neurons with LY294002 a specific PI3K inhibitor, insulin-induced increases in spine formation could be inhibited, resulting in complete halt of insulin-induced increase in spine formation. Also, by examining whether a target of Akt, mTOR was a factor in insulin-induced increase in spine density, it was concluded that insulin-induced increase in spine density is associated with activation of the PI3K/Akt/mTOR signaling pathway (Lee, Huang and Hsu, 2011).
In a study published by Gabbouj et al (2019), 14-month-old WT and APPSwe/PS1dE9 (APP/PS1) transgenic mice were used to investigate how a single dose of intranasal insulin modulates brain glucose metabolism by FDG-PET and effect on spatial learning and memory. How insulin influences the activity of Akt1 and Akt2 kinases, expression of glial and neuronal markers, and autophagy in the hippocampus was also assessed. It was demonstrated that intranasal insulin treatment increased glucose metabolism in the ventral brain areas and hippocampus of WT mice, but no increase was detected in APP/PS1 mice (Gabbouj et al., 2019).
Intranasal insulin treatment specifically activated Akt2 and its downstream signaling in the hippocampus of WT, but not APP/PS1 mice. Insulin differentially affected the expression of homeostatic microglia markers P2ry12 and Cx3cr1 and autophagy in the hippocampus of WT and APP/PS1 mice (Gabbouj et al., 2019). Gabbouj et al. (2019) concluded that intranasal insulin exerts diverse effects on autophagy, homeostatic status of microglia and Akt2 signaling, such as decreased transcriptional activity of FoxO1 consistent with Latva-Rasku et al. (2018).
One week of daily intranasal insulin led to a 50% reduction of the Aβ40 levels in the forebrains of 9-month-old female 3xTg-AD mice (Chen et al., 2014). Further implicating that intranasal insulin may hinder amyloidogenesis in AD, treatment of female 4.5- month-old APP/PS1 mice for 6 weeks with daily intranasal insulin reduced amyloid plaques in both the cortex and hippocampus, and lowered concentrations of soluble Aβ oligomers in the brain (Mao et al., 2016).
As animal studies indicate, intranasal insulin may exert ameliorating effects on additional neuropathological processes, hindering the development and progression of AD. For instance, 9-month-old 3xTg-AD mice treated with a daily dose of intranasal insulin for one week exhibited signs of reduced microglia activation and increased levels of synaptic proteins (Chen et al., 2014). However, repeated doses of intranasal insulin in 21-month-old male rats resulted in glial overactivation (Anderson et al., 2017), implying that further research on interplay between insulin and microglia may be necessary.
Another animal study has shown that long-term intranasal insulin treatment may have tau hyperphosphorylation ameliorating effect through PI3K/Akt signaling pathway. The 6-week intranasal insulin treatment to ICV-STZ treated rats resulted in no changes in blood glucose but ameliorated the memory deficit effects. Intranasal insulin treatment resulted in changes in the total and phosphorylated levels of insulin receptor β-subunit (IRβ), insulin receptor substrate-1 (IRS1), PI3K, PDK1 and Akt, compared to control rats. Intranasal insulin treatment decreased the phosphorylation of tau, upregulated inhibitory phosphorylation of GSK3β at S9 residue and GSK3α at S21 residue in the hippocampus of ICV-STZ rats (Chen et al., 2018).
Recent studies suggest that a single dose of intranasal insulin is already sufficient to improve cognitive performance in cognitively healthy humans (Benedict et al., 2008; Novak et al., 2014; Brünner et al., 2015). For instance, a single intranasal dose of insulin improved visuospatial memory and verbal fluency functions in a sample of elderly non-demented T2D patients and aged-matched subjects (Novak et al., 2014). However, this may not apply in every case. For instance, in cognitively healthy subjects aged older than 70 years used as controls for patients with mild cognitive impairment and AD, a single dose of intranasal insulin (10 to 60 IU) failed to improve attention, working memory, and story recall (Reger et al., 2006).
In a 4-month trial of regular intranasal insulin therapy (20 IU; 40 IU; and a placebo daily) showed reduced progression of hypometabolism in the parietotemporal, frontal, precuneus, and cuneus brain regions in those treated with insulin (Craft et al., 2012). In a smaller trial intranasal administration of regular insulin (40 IU daily for four months) improved memory and preserved brain volume (Craft et al., 2017), confirming long-term neuroprotective effects of insulin.
Due to the long window of preclinical AD (Jack et al., 2013), it may be important to investigate in cognitively healthy humans whether intranasal insulin influences tau metabolism and turnover of Aβ in the circulation or in CSF. Additionally, gender, APOE genotype, and the type of insulin formulation appear to considerably modify patient response. It has been suggested that studies with longer durations and specific sample populations are needed to paint a clearer picture of the connection between insulin and AD pathology (Chapman et al., 2018).
5 Discussion¶
PI3K/Akt signaling pathway exists in various organs in the body and plays an important role in a variety of physiological functions, such as brain. Also, PI3K/Akt signaling pathway plays a key role in development and progression of AD. Here we describe the metabolic and inflammatory functions of PI3K/Akt signaling pathway in the brain and its relationship with T2D and the effects that result from dysfunction of PI3K/Akt signaling pathway in the brain and its link with AD. There are several factors that alter of PI3K/Akt signaling pathway in the brain, such as inflammatory response induced by activated microglia, accumulation of Aβ aggregates in synaptic clefts, intra- and extraneuronal NFTs in the brain, impaired insulin signaling in the brain and mutations of Akt2. Intricate relationships between these factors remain to be elucidated, for there still exist contradicting evidence, such as with the case of Aβ (Ali and Kim, 2015; Singh et al., 2017). Evidence from epidemiological, clinical and animal studies has established strong links between T2D and neurodegeneration such as that observed in AD, although all the intricate mechanisms that link these together remain to be determined. As the population ages, the prevalence of dementia increases. Much of this increase will take place in low and middle income countries, most notably in Asia and cost of dementia in 2015 was estimated to be US$ 818 billion (Prince et al., 2015). Additionally prevalence of diabetes is growing world-wide, especially in developing countries (Wild et al., 2004). Thus, as the purchasing power of people in emerging markets grow, it may be necessary to consider the cost benefit of early intervention on consumer choice and investment in antidiabetic treatment. Through prevention diabetes, also the global increase in AD cases could be ameliorated by preventing dysfunctional insulin signaling in brain.
6 Sources¶
- Ali, T. and Kim, M. O. (2015) ‘Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3β pathway in the mouse hippocampus’, Journal of Pineal Research, 59(1), pp. 47–59. doi: 10.1111/jpi.12238.
- Apelt, J., Mehlhorn, G. & Schliebs, R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 57, 693–705 (1999).
- Anderson, K. L. et al. (2017) ‘Impact of Single or Repeated Dose Intranasal Zinc-free Insulin in Young and Aged F344 Rats on Cognition, Signaling, and Brain Metabolism’, The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 72(2), pp. 189–197. doi: 10.1093/gerona/glw065.
- Arnold, S. E. et al. (2018) ‘Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums’, Nature Reviews Neurology. Nature Publishing Group, 14(3), pp. 168–181. doi: 10.1038/nrneurol.2017.185.
- Asai, H. et al. (2015) ‘Depletion of microglia and inhibition of exosome synthesis halt tau propagation’, Nature Neuroscience. Nature Publishing Group, 18(11), pp. 1584–1593. doi: 10.1038/nn.4132.
- Bedse G, Domenico FD, Serviddio G, Cassano T (2015) Aberrant insulin signaling in Alzheimer's disease: current knowledge, Frontiers in Neuroscience, 9:204, doi: 10.3389/fnins.2015.00204.
- Benedict, C. et al. (2008) ‘Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin’, Journal of Clinical Endocrinology and Metabolism, 93(4), pp. 1339–1344. doi: 10.1210/jc.2007-2606.
- Brünner, Y. F. et al. (2015) ‘Central insulin administration improves odor cued reactivation of spatial memory in young men’, Journal of Clinical Endocrinology and Metabolism, 100(1), pp. 212–219. doi: 10.1210/jc.2014-3018.
- Chapman, C. D. et al. (2018) ‘Intranasal insulin in Alzheimer’s disease: Food for thought’, Neuropharmacology, 136, pp. 196–201. doi: 10.1016/j.neuropharm.2017.11.037.
- Chen, Y. et al. (2014) ‘Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Aβ level and microglia activation in the brains of 3xTg-AD mice’, Experimental Neurology. Elsevier B.V., 261, pp. 610–619. doi: 10.1016/j.expneurol.2014.06.004.
- Chen, Y. et al. (2018) ‘Intranasal Insulin Ameliorates Cerebral Hypometabolism, Neuronal Loss, and Astrogliosis in Streptozotocin-Induced Alzheimer’s Rat Model’, Neurotoxicity Research. Neurotoxicity Research, 33(4), pp. 716–724. doi: 10.1007/s12640-017-9809-7.
- Cohen, M. M. (2013) ‘The AKT genes and their roles in various disorders’, American Journal of Medical Genetics, Part A, 161(12), pp. 2931–2937. doi: 10.1002/ajmg.a.36101.
- Cooper M. and Hausman (2009) ‘Pathways of Intracellular Signal Transduction’ in The Cell, 6th edn. Sinauer Associates Inc. pp. 608-628, ISBN:9780878939640.
- Craft, S. et al. (2012) ‘Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial’, Archives of Neurology, 69(1), pp. 29–38. doi: 10.1001/archneurol.2011.233.
- Craft, S. et al. (2017) ‘Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial’, Journal of Alzheimer’s Disease, 57(4), pp. 1325–1334. doi: 10.3233/JAD-161256.
- Deczkowska, A. et al. (2018) ‘Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration’, Cell. Elsevier Inc., 173(5), pp. 1073–1081. doi: 10.1016/j.cell.2018.05.003.
- de la Monte, S. M. (2017). Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer's disease. Drugs 77, 47–65. doi: 10.1007/s40265-016-0674-0.
- Fruman, D. A., Meyers, R. E. and Cantley, L. C. (1998) ‘Phosphoinositide Kinases’, Annual Review of Biochemistry, 67(1), pp. 481–507. doi: 10.1146/annurev.biochem.67.1.481.
- Gabbouj, S. et al. (2019) ‘Intranasal insulin activates Akt2 signaling pathway in the hippocampus of wild-type but not in APP/PS1 Alzheimer model mice’, Neurobiology of Aging, 75. doi: 10.1016/j.neurobiolaging.2018.11.008.
- Gong, C.-X. and Iqbal, K. (2008) ‘Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease’, Current Medicinal Chemistry, 15(23), pp. 2321–2328. doi: 10.2174/092986708785909111.
- Hawkins, P. T. and Stephens, L. R. (2015) ‘PI3K signalling in inflammation’, Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids, 1851(6), pp. 882–897. doi: 10.1016/j.bbalip.2014.12.006.
- Henriksen EJ, Diamond SMK, Marchionne EM. 2011. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 51:993–999.
- Hensley, K. and Kursula, P. (2016) ‘Collapsin Response Mediator Protein-2 (CRMP2) is a Plausible Etiological Factor and Potential Therapeutic Target in Alzheimer’s Disease: Comparison and Contrast with Microtubule-Associated Protein Tau’, Journal of Alzheimer’s Disease, 53(1), pp. 1–14. doi: 10.3233/JAD-160076.
- Iqbal, K., Liu, F. and Gong, C. X. (2016) ‘Tau and neurodegenerative disease: The story so far’, Nature Reviews Neurology, 12(1), pp. 15–27. doi: 10.1038/nrneurol.2015.225.
- Jack, C. R. et al. (2013) ‘Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers’, The Lancet Neurology, 12(2), pp. 207–216. doi: 10.1016/S1474-4422(12)70291-0.
- Jay, T.R., Hirsch, A.M., Broihier, M.L., Miller, C.M., Neilson, L.E., Ransohoff, R.M., Lamb, B.T., and Landreth, G.E., Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37, 637–647 (2017).
- Kang, J. and Muller-Hill, B. (1990) ‘Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: PreA4695 mRNA is predominantly produced in rat and human brain’, Biochemical and Biophysical Research Communications, vol. 166, no. 3, pp. 1192–1200.
- Kang, Q. et al. (2017) ‘MiR-124-3p attenuates hyperphosphorylation of Tau protein- induced apoptosis via caveolin-1-PI3K / Akt / GSK3β pathway in’, 8(15), pp. 24314–24326.
- Kothari, V. et al. (2017) ‘High fat diet induces brain insulin resistance and cognitive impairment in mice’, Biochimica et Biophysica Acta - Molecular Basis of Disease. Elsevier B.V., 1863(2), pp. 499–508. doi: 10.1016/j.bbadis.2016.10.006.
- Latva-Rasku, A. et al. (2018) ‘A partial loss-of-function variant in AKT2 is associated with reduced insulin-mediated glucose uptake in multiple insulin-sensitive tissues: A genotype-based callback positron emission tomography study’, Diabetes, 67(2), pp. 334–342. doi: 10.2337/db17-1142.
- Laws, S. M. et al. (2017) ‘Insulin resistance is associated with reductions in specific cognitive domains and increases in CSF tau in cognitively normal adults’, Scientific Reports, 7(1), pp. 1–11. doi: 10.1038/s41598-017-09577-4.
- Lee, C. C., Huang, C. C. and Hsu, K. Sen (2011) ‘Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways’, Neuropharmacology. Elsevier Ltd, 61(4), pp. 867–879. doi: 10.1016/j.neuropharm.2011.06.003.
- Leibrock, C. et al. (2013) ‘Akt2 deficiency is associated with anxiety and depressive behavior in mice’, Cellular Physiology and Biochemistry, 32(3), pp. 766–777. doi: 10.1159/000354478.
- Levenga, J. et al. (2017) ‘AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity’, eLife, 6, pp. 1–24. doi: 10.7554/eLife.30640.
- Li, L. and Hölscher, C. (2007) ‘Common pathological processes in Alzheimer disease and type 2 diabetes: A review’, Brain Research Reviews, 56(2), pp. 384–402. doi: 10.1016/j.brainresrev.2007.09.001.
- Lodish, H. et al. (2016) ‘Phosphoinositide Signaling Pathways’, in Molecular Cell Biology. 8th edn. Katherine Ahr Parker, pp. 748–751.
- Mao, Y. F. et al. (2016) ‘Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice’, Aging Cell, 15(5), pp. 893–902. doi: 10.1111/acel.12498.
- Moll, L. and Schubert, M. (2012) ‘The role of insulin and insulin-like growth factor-1/FoxO-mediated transcription for the pathogenesis of obesity-associated dementia’, Current Gerontology and Geriatrics Research, 2012. doi: 10.1155/2012/384094.
- Mehlhorn, G., Hollborn, M., & Schliebs, R. (2000). Induction of cytokines in glial cells surrounding cortical β-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. International Journal of Developmental Neuroscience, 18(4-5), 423–431.doi:10.1016/s0736-5748(00)00012-5
- Noguchi, M. and Suizu, F. (2012) ‘Regulation of AKT by Phosphorylation of Distinct Threonine and Serine Residues Phosphorylation Controls the Intracellular Signal Transduction’, 47.
- Novak, V. et al. (2014) ‘Enhancement of vasoreactivity andcognition by intranasal insulin in Type 2 diabetes’, Diabetes Care, 37(3), pp. 751–759. doi: 10.2337/dc13-1672.
- Pearson, H. A. and Peers, C. (2006) ‘Physiological roles for amyloid β peptides’, Journal of Physiology, 575(1), pp. 5–10. doi: 10.1113/jphysiol.2006.111203.
- Prince, M. et al. (2015) ‘World Alzheimer Report 2015 The Global Impact of Dementia An analysis of prevalence, incidence, cost and trends’, Alzheimer’s Disease International. doi: 10.1111/j.0963-7214.2004.00293.x.
- Rangaraju, S. et al. (2018) ‘Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease’, Molecular Neurodegeneration. Molecular Neurodegeneration, 13(1), pp. 1–25. doi: 10.1186/s13024-018-0254-8.
- Reger, M. A. et al. (2006) ‘Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype’, Neurobiology of Aging, 27(3), pp. 451–458. doi: 10.1016/j.neurobiolaging.2005.03.016.
- Reinhard, C., Hébert, S. S. and De Strooper, B. (2005) ‘The amyloid-?? precursor protein: Integrating structure with biological function’, EMBO Journal, 24(23), pp. 3996–4006. doi: 10.1038/sj.emboj.7600860.
- Reitz, C. (2012) ‘Alzheimer’s disease and the amyloid cascade hypothesis: A critical review’, International Journal of Alzheimer’s Disease, 2012. doi: 10.1155/2012/369808.
- Serrano-Pozo, A. et al. (2011) ‘Neuropathological alterations in Alzheimer disease’, Cold Spring Harbor Perspectives in Medicine, 1(1), pp. 1–23. doi: 10.1101/cshperspect.a006189.
- Singh, A. K. et al. (2017) ‘Neuroprotection Through Rapamycin-Induced Activation of Autophagy and PI3K/Akt1/mTOR/CREB Signaling Against Amyloid-β-Induced Oxidative Stress, Synaptic/Neurotransmission Dysfunction, and Neurodegeneration in Adult Rats’, Molecular Neurobiology. Molecular Neurobiology, 54(8), pp. 5815–5828. doi: 10.1007/s12035-016-0129-3.
- Spielman, L. J., Bahniwal, M., Little, J. P., Walker, D. G. & Klegeris, A. Insulin modulates in vitro secretion of cytokines and cytotoxins by human glial cells. Curr. Alzheimer Res. 12, 684–693 (2015).
- Thotala, D. and Yazlovitskaya, E. (2011) ‘GSK3B (glycogen synthase kinase 3 beta)’, Atlas of Genetics and Cytogenetics in Oncology and Haematology, 12(1), pp. 7–10. doi: 10.4267/2042/44931.
- Vanhaesebroeck, B. et al. (2010) ‘The emerging mechanisms of isoform-specific PI3K signalling’, Media International Australia. Nature Publishing Group, 11(139), pp. 124–132. doi: 10.1038/nrm2882.
- Vassar, R. (2004) ‘The β-Secretase Enzyme in Alzheimer’s Disease’, Molecular Biology, 23, pp. 105–113. Available at: https://link.springer.com/content/pdf/10.1385%2FJMN%3A23%3A1-2%3A105.pdf.
- Verdile, G. et al. (2015) ‘Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease’, Mediators of Inflammation. Hindawi Publishing Corporation, 2015. doi: 10.1155/2015/105828. Wild, S. et al. (2004) ‘Estimates for the year 2000 and projections for 2030’, Diabetes care, 27(5), pp. 1047–1053. doi: ISBN 92 4 159493 4.
- Yarchoan, M. et al. (2014) ‘Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies’, Acta Neuropathologica, 128(5), pp. 679–689. doi: 10.1007/s00401-014-1328-5.
- Ye, P., Li, L., Lund, P. K. & D’Ercole, A. J. Deficient expression of insulin receptor substrate 1 (IRS 1) fails to block insulin-like growth factor I (IGF I) stimulation of brain growth and myelination. Brain Res. Dev. Brain Res. 136, 111–121 (2002).
- Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016 Sep 1;143(17):3050-60. doi: 10.1242/dev.137075. PMID: 27578176.
- Zheng, H. et al. (2017) ‘TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway’, The Journal of Neuroscience, 37(7), pp. 1772–1784. doi: 10.1523/JNEUROSCI.2459-16.2017.
